Whole-Genome DNA Methylation data analysis pipeline
From sequencing data to differential methylation analysis
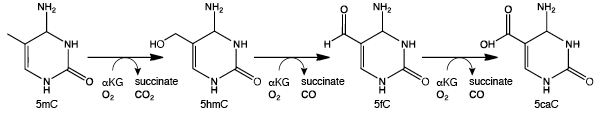
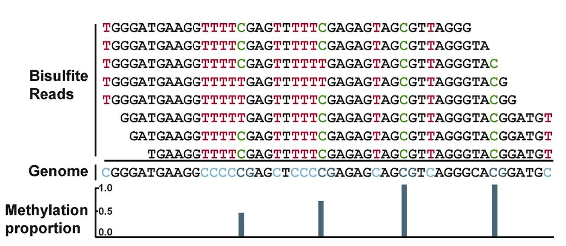
A brief overview of DNA Methylation
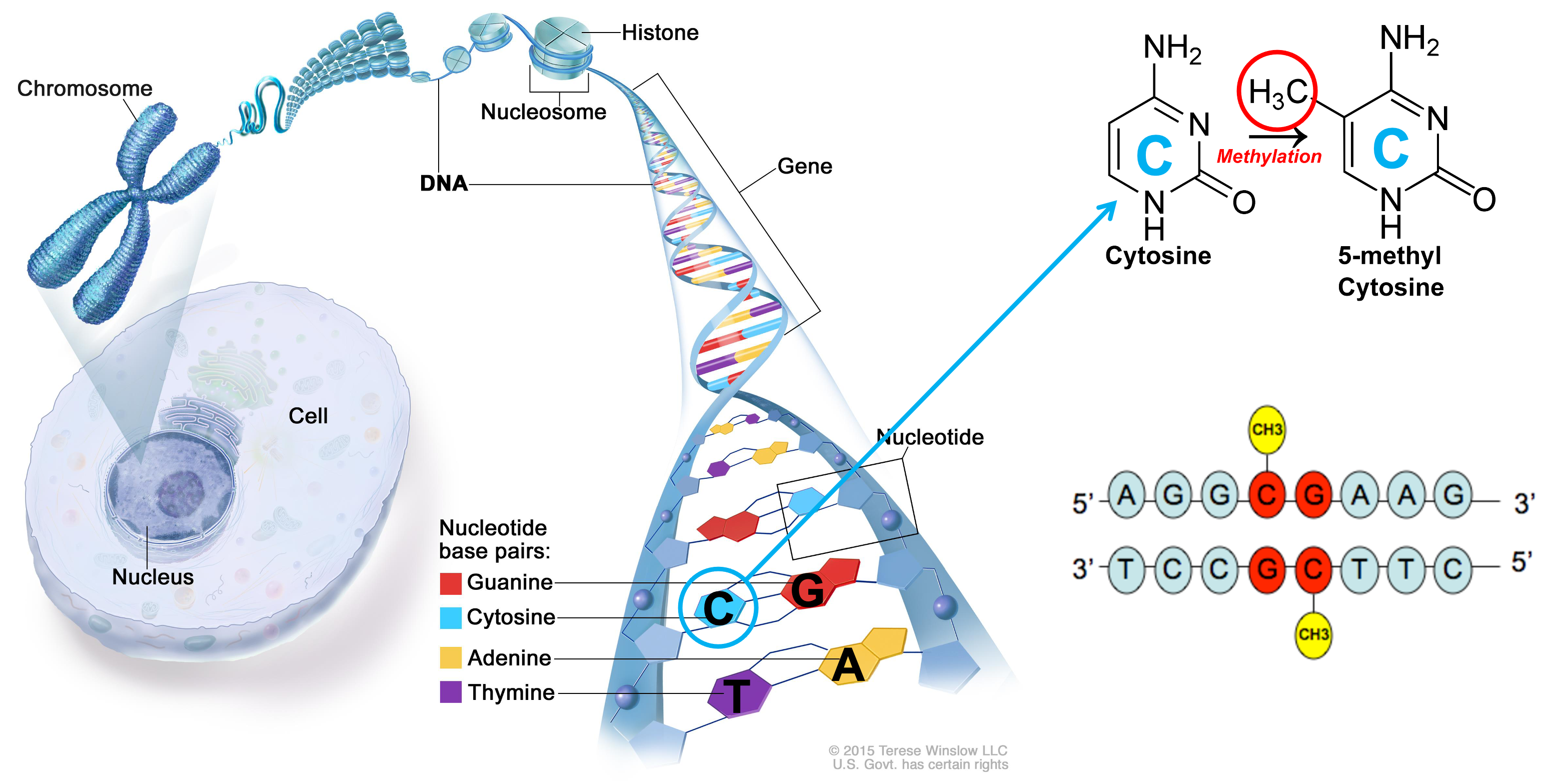

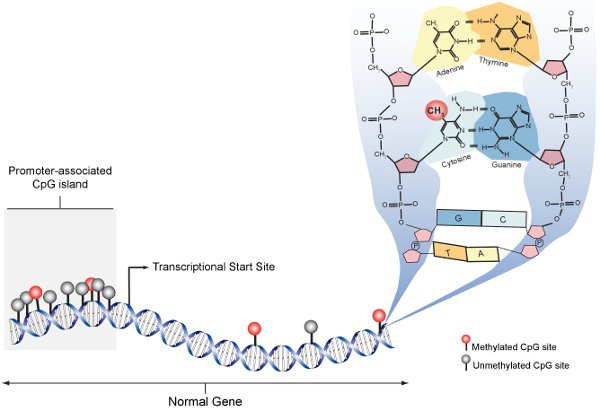
- The human genome contains ~32 million CpGs. VERY different between different species
- CpGs are not evenly spaced across the genome. Tend to be present in clusters, CpG islands
- CpG methylation is spatially correlated
Detection of DNA Methylation
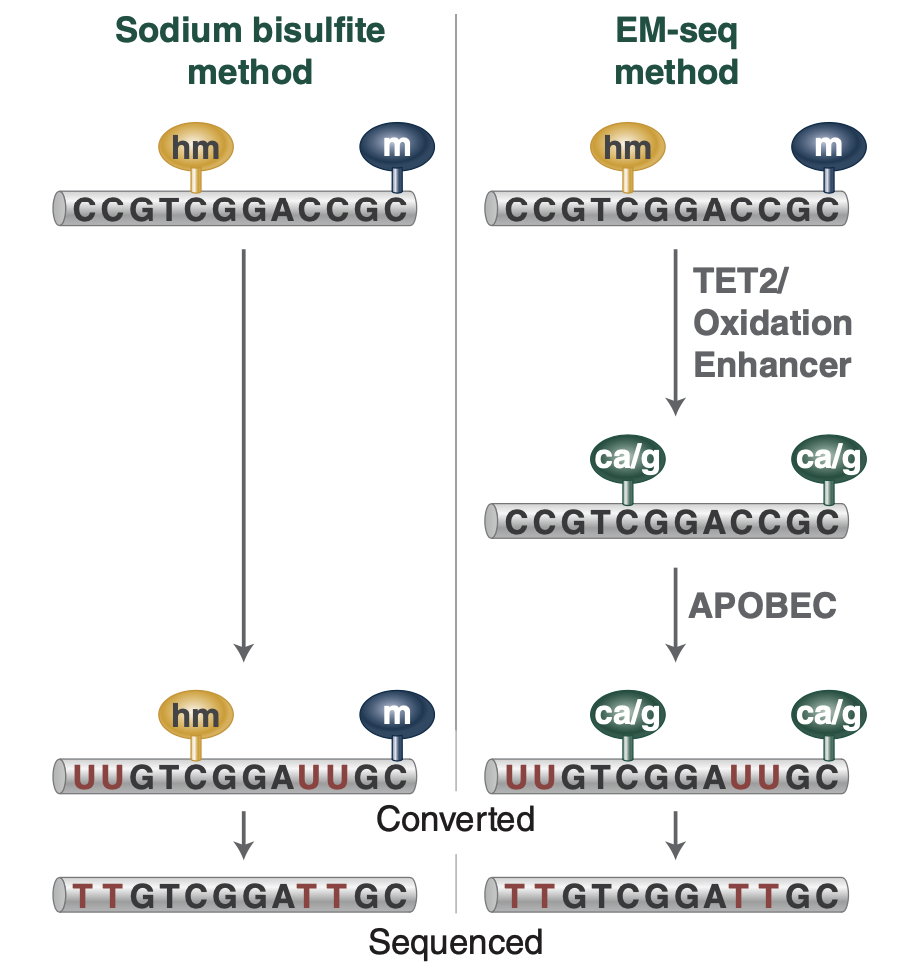
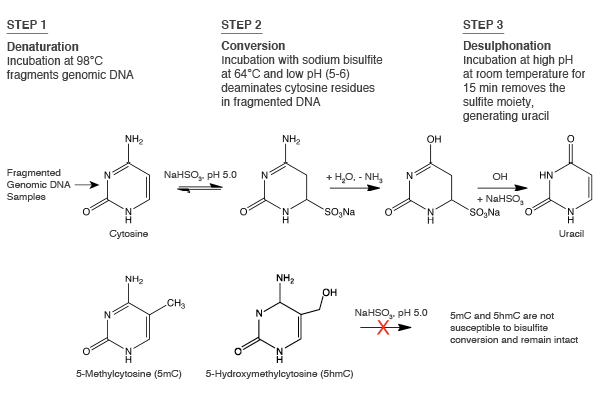
Why Twist?
Highly Sensitive Detection of Differentially Methylated Regions
NEBNext® EM-seqTM Kit for Twist Targeted Methylation Sequencing
Twist Methylation Enhancer
Twist Fast Hybridization System and Custom Methylation Panels
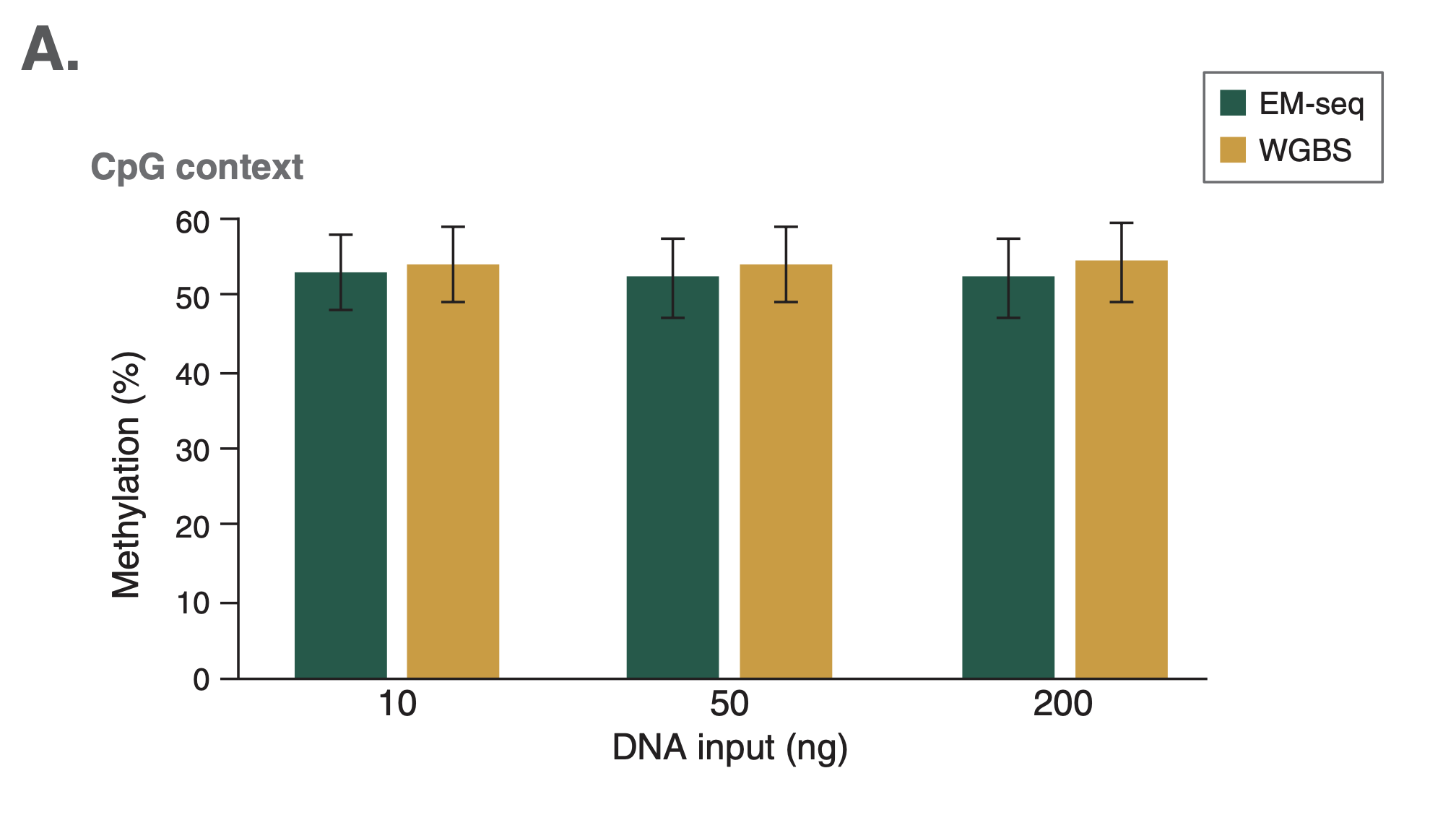
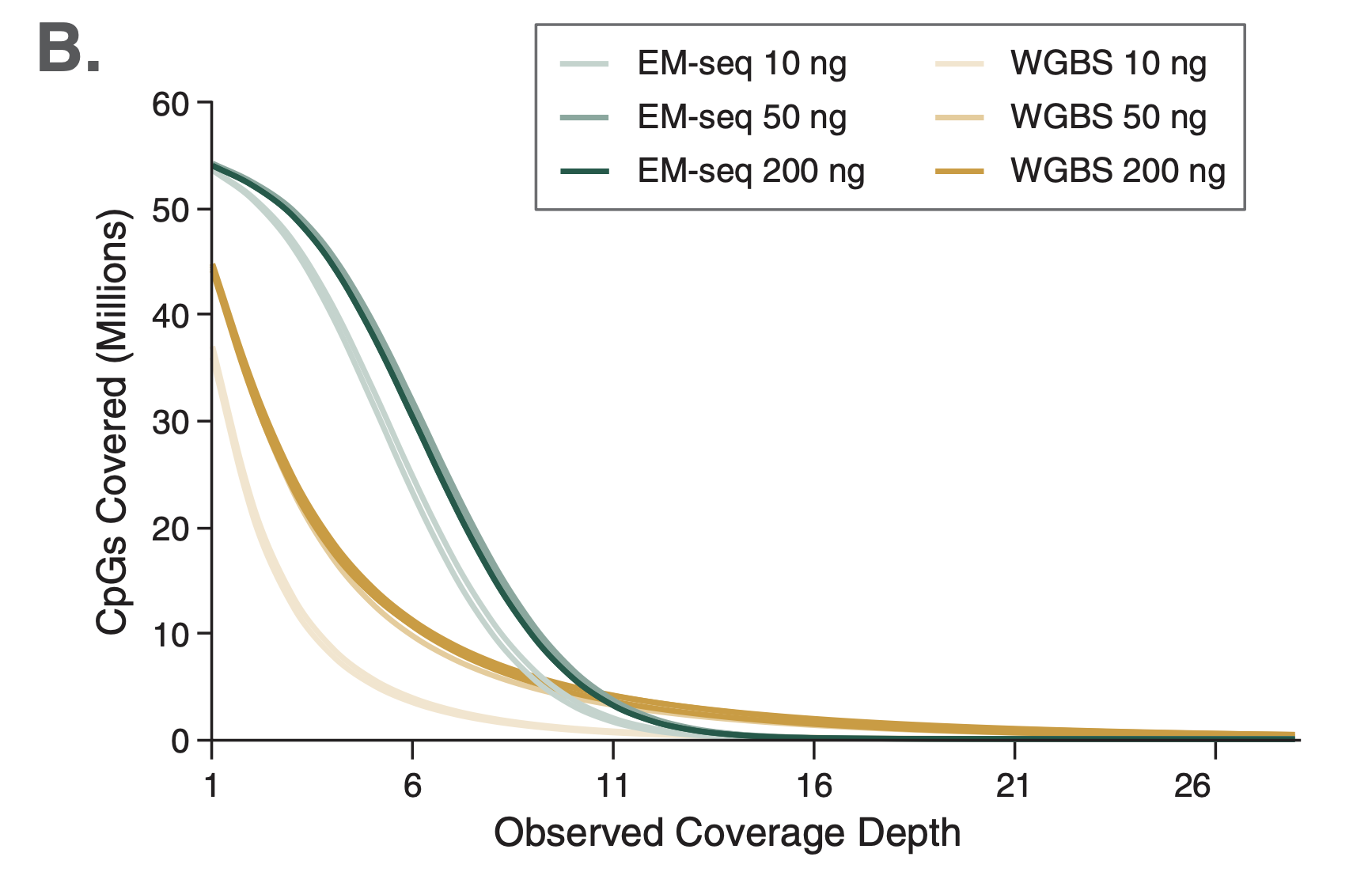
Comparison among technologies
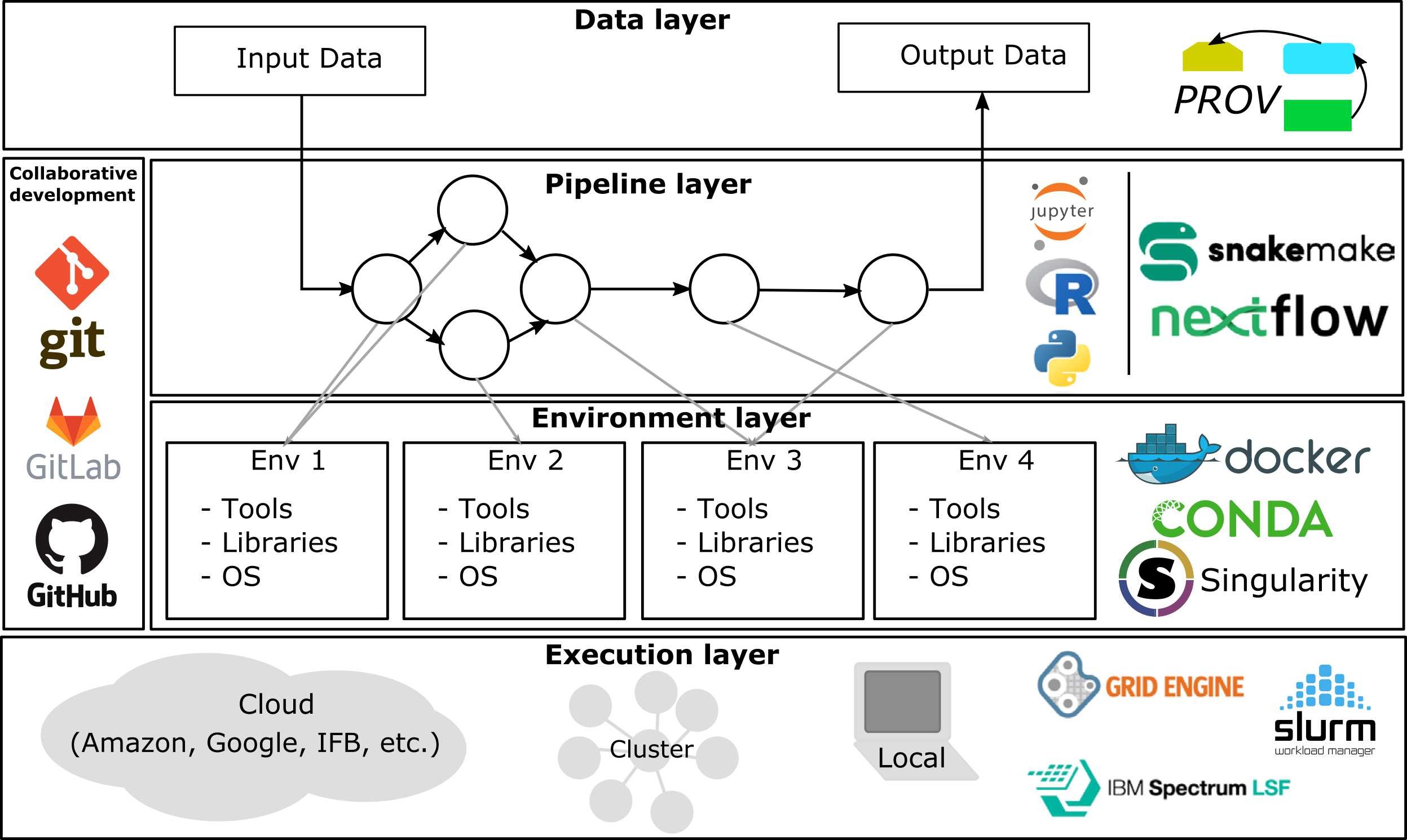
Bioinformatics pipelines
A bioinformatics pipeline typically depends on the availability of several resources, including adequate storage, computer units, network connectivity, and appropriate software execution environment.
Automation
Containers
Version control
Clinical validation
Twist-suggested DNA methylation data analysis
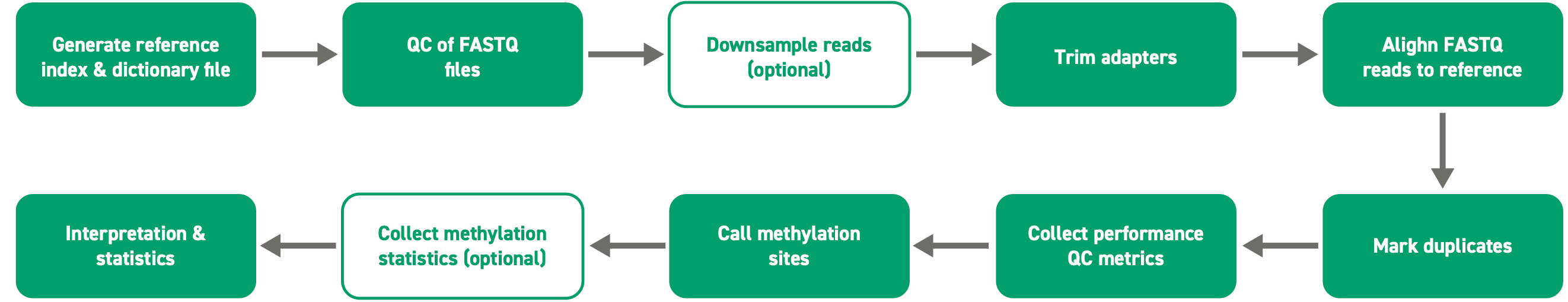
Optional steps:
- Downsampling reads (seqtk)
- Collect methylation stats
Important information
1. same pipeline can be used for Twist custom methylation panel
2. genome reference file should match the panel’s genome build
3. replace the Methylome V1 probe and target BED file.
Tools for methylation data processing

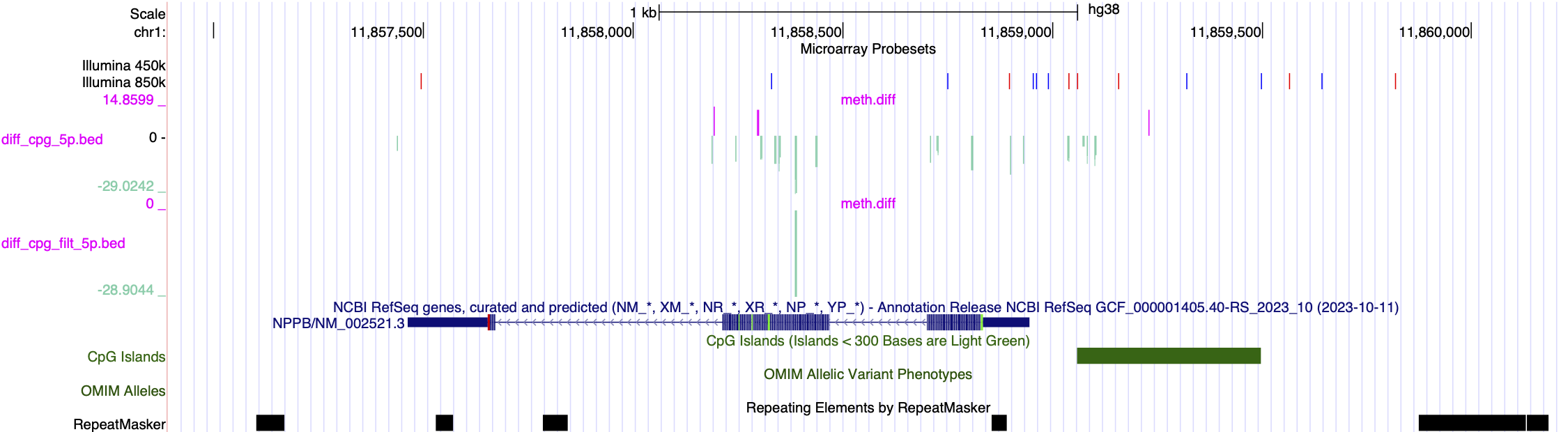
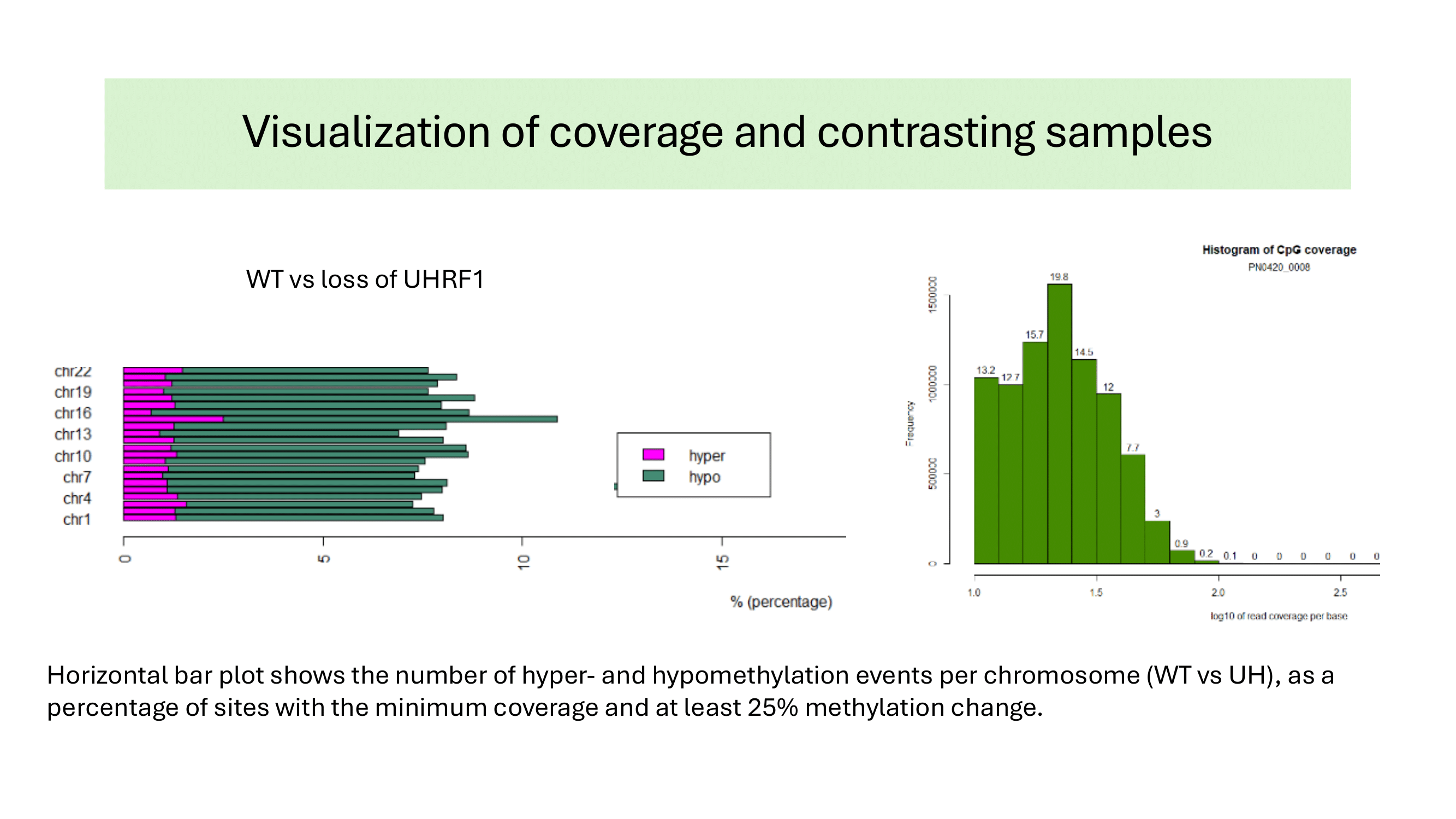
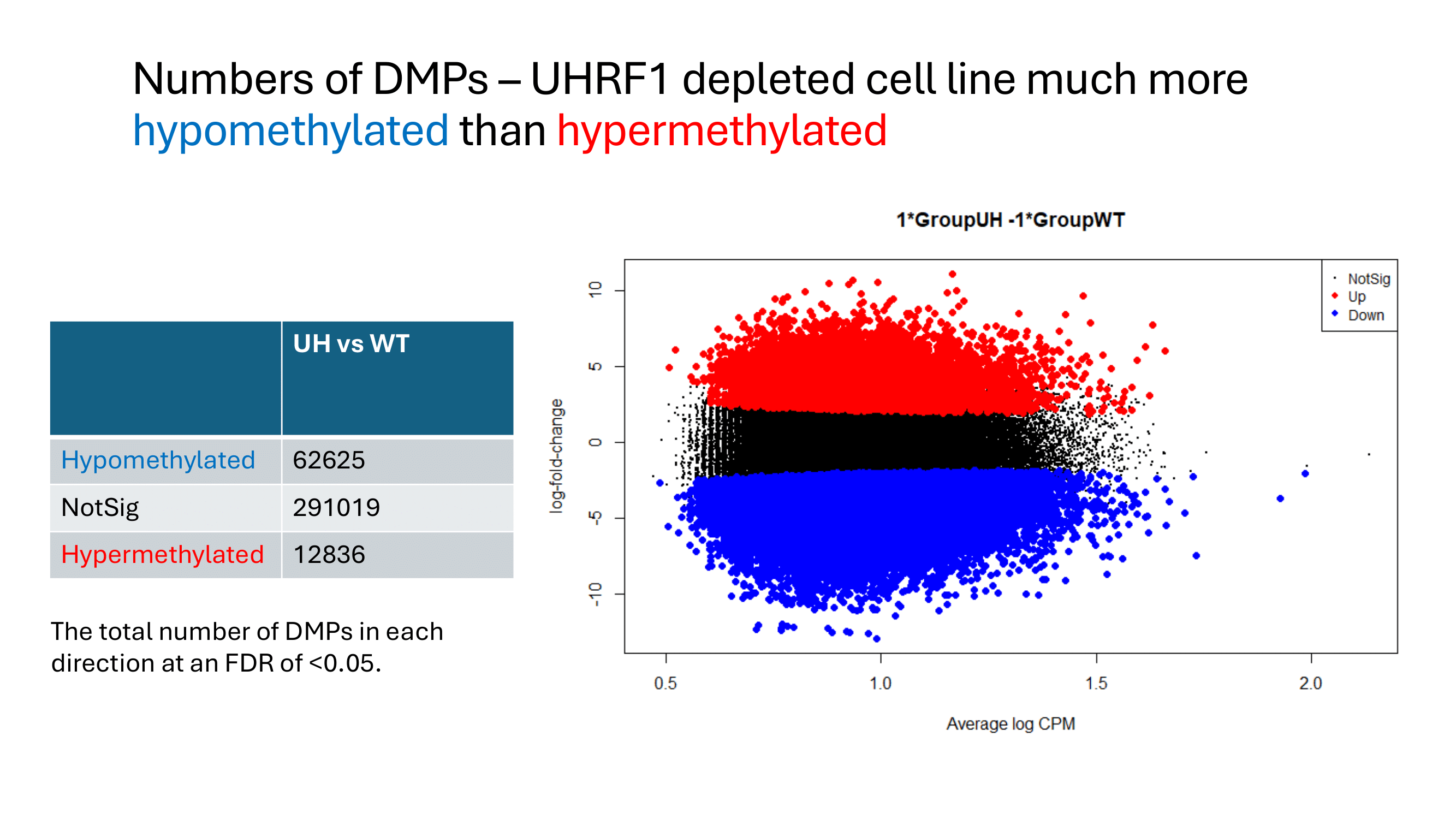
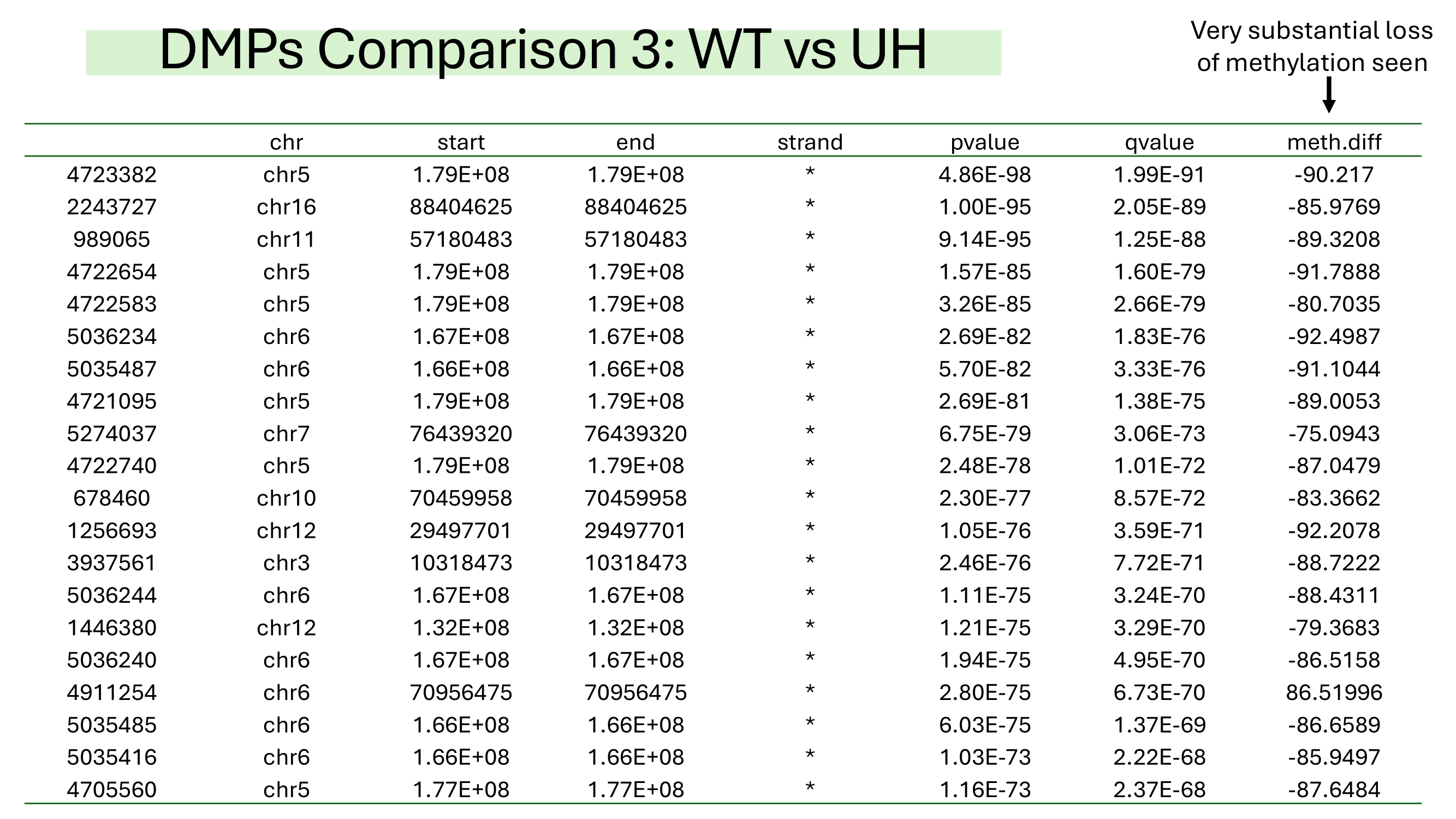
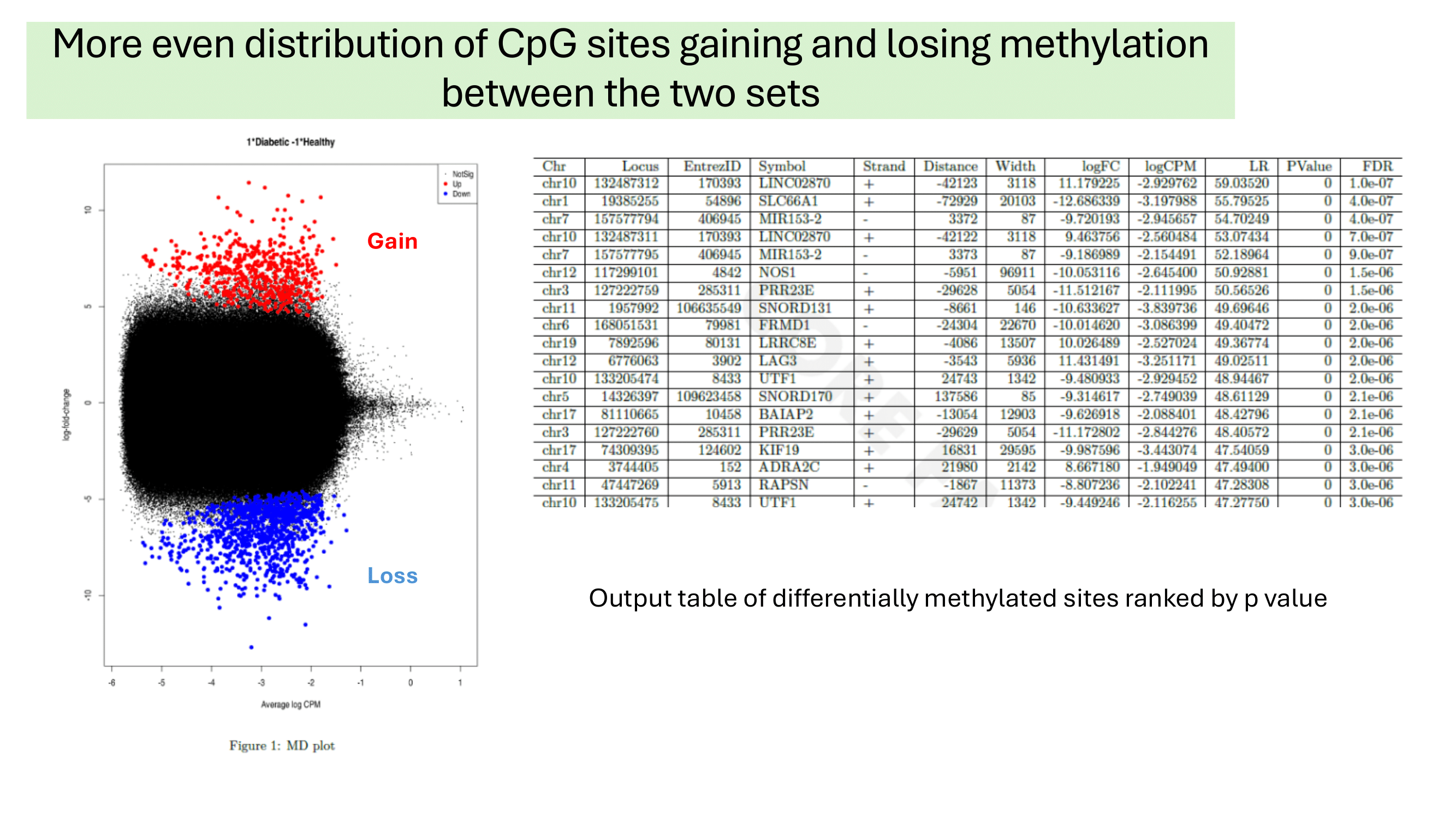
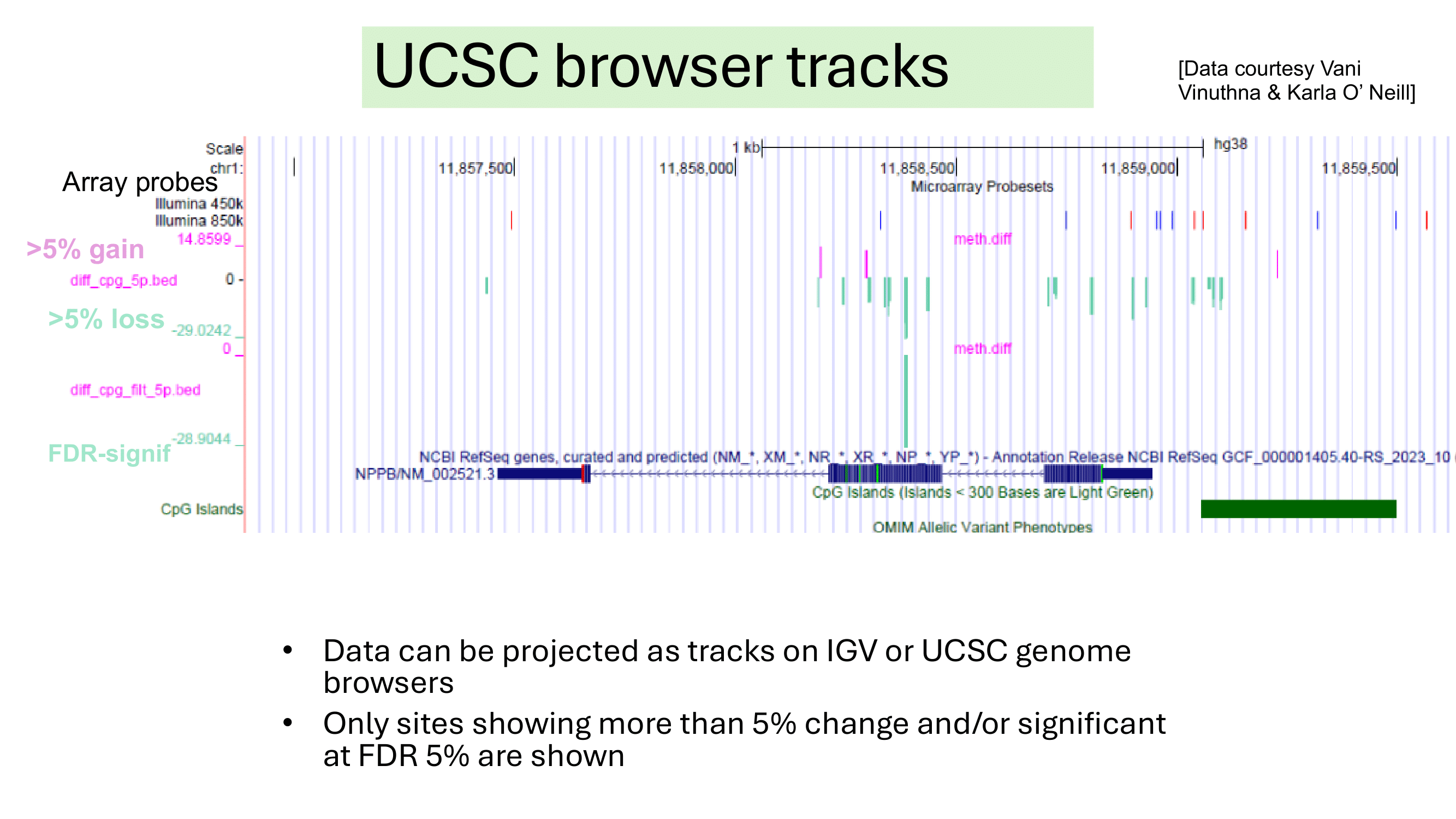
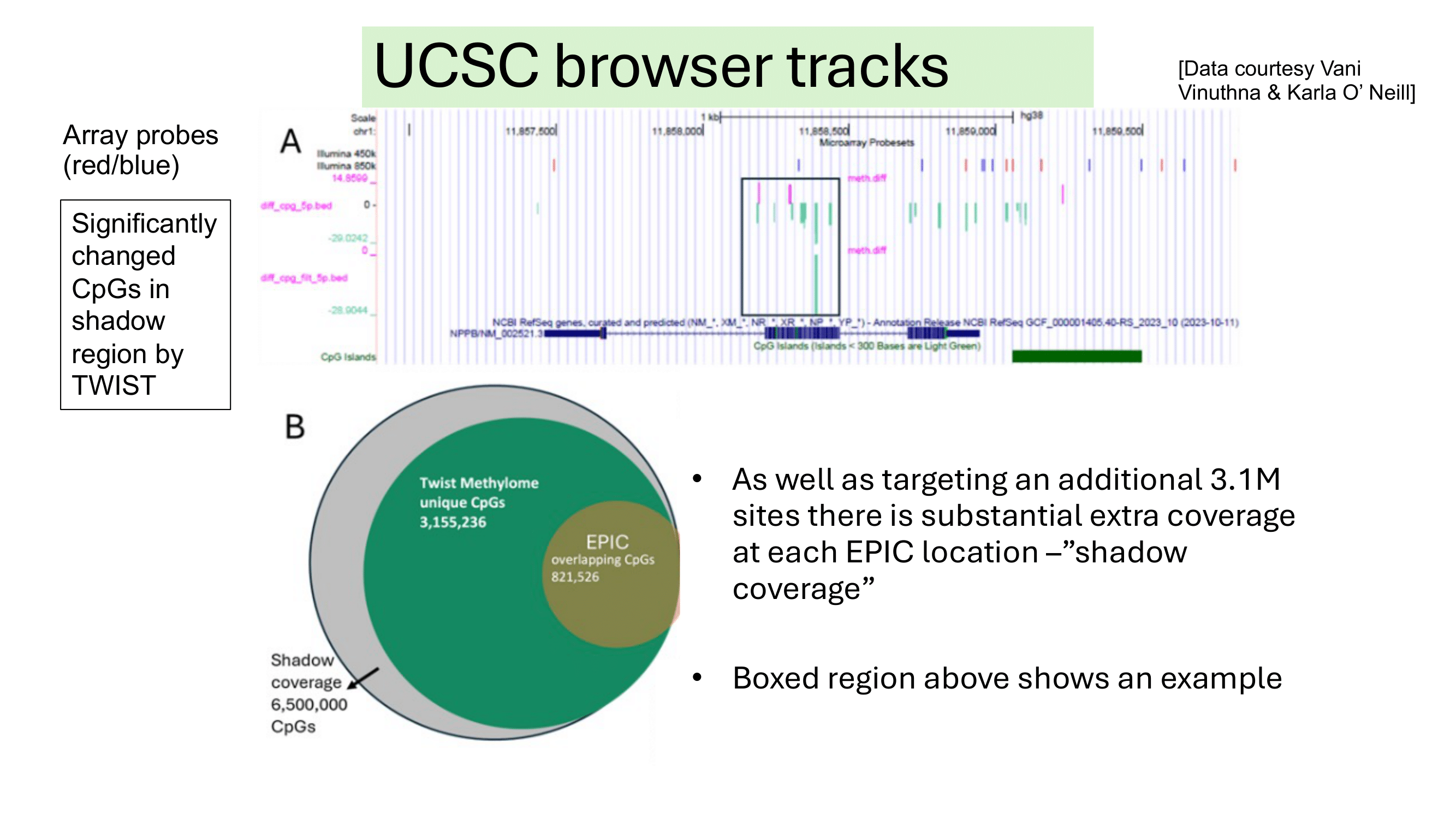
Some examples of methylation data analysis results
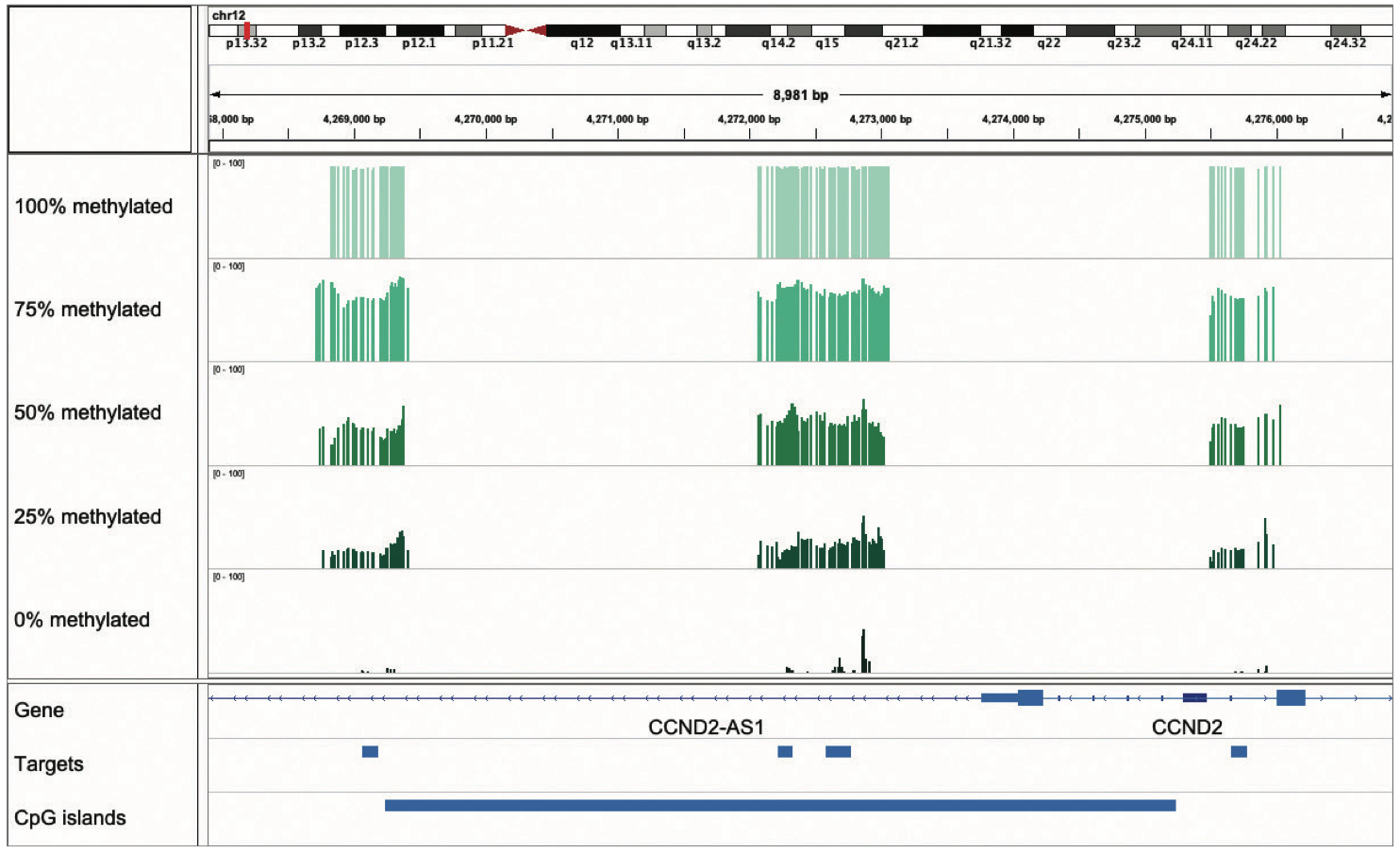
Genome Browser View showing region chr1:11,857,464-11,858,945 of the hg38 assembly. 1
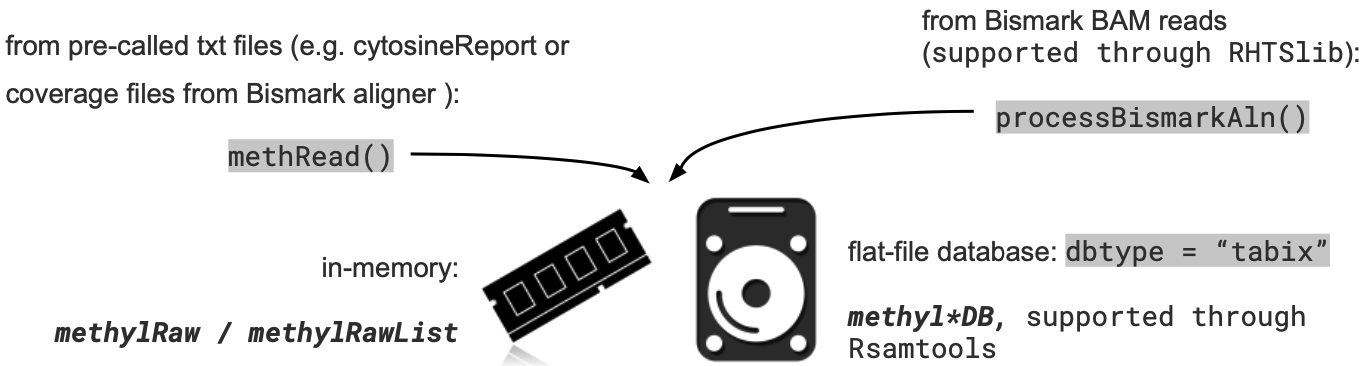
A1. MethylKit: Reading data

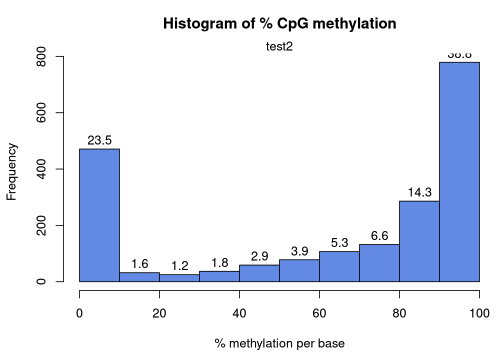
A2. MethylKit: Summarize statistics on samples
getMethylationStats(methylRaw) methylation statistics per base
summary:
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00 20.00 82.79 63.17 94.74 100.00
percentiles:
0% 10% 20% 30% 40% 50% 60% 70%
0.00000 0.00000 0.00000 48.38710 70.00000 82.78556 90.00000 93.33333
80% 90% 95% 99% 99.5% 99.9% 100%
96.42857 100.00000 100.00000 100.00000 100.00000 100.00000 100.00000
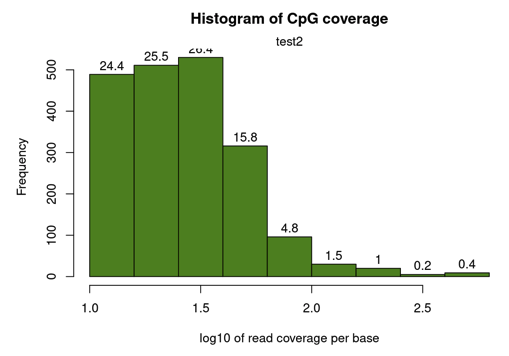
getCoverageStats(methylRaw)read coverage statistics per base
summary:
Min. 1st Qu. Median Mean 3rd Qu. Max.
10.00 16.00 26.00 34.45 39.00 630.00
percentiles:
0% 10% 20% 30% 40% 50% 60% 70%
10.000 11.000 14.000 17.000 20.000 26.000 30.000 36.000
80% 90% 95% 99% 99.5% 99.9% 100%
42.000 60.000 78.750 195.800 328.300 441.945 630.000
A3. MethylKit: Compare Samples
get the bases covered in all samples: merge all samples to one object for base-pair locations that are covered in all samples:
assocComp - Batch Effect correction
tileMethylCounts - Tiling window analysis
unite(methylRawList) --> methylBase
getCorrelation(methylBase)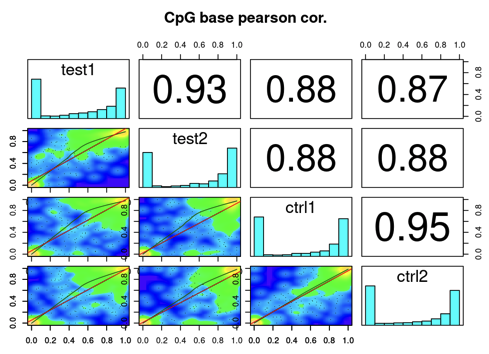
clusterSamples(methylBase)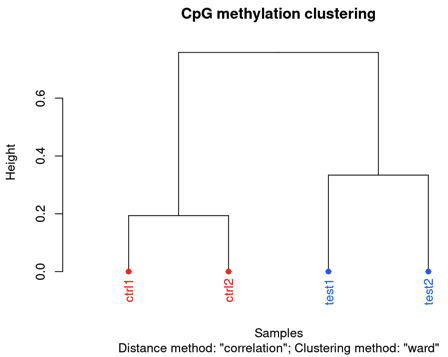
PCASamples(methylBase)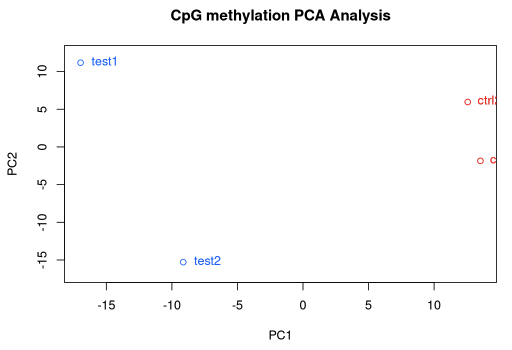
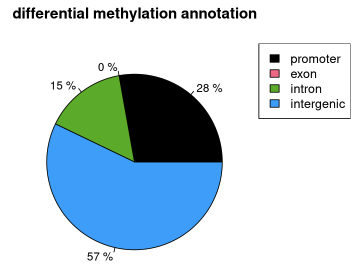
A5. MethylKit: Annotation
Use genomation package to annotate differentially methylated regions/bases based on gene annotation:
first read the gene BED file:
gene.obj=readTranscriptFeatures(system.file("extdata", "refseq.hg18.bed.txt",package = "methylKit"))
then get all differentially methylated bases:
myDiff25p=getMethylDiff(methylDiff,difference=25, qvalue=0.01)now annotate differentially methylated CpGs with promoter/exon/intron using annotation data
diffAnn=annotateWithGeneParts(as(myDiff25p,"GRanges"), gene.obj)
finally visualize the annotation:
plotTargetAnnotation(diffAnn,precedence=TRUE, main="differential methylation annotation")![]()
B4. EdgeR: Calculating differential methylation
Dispersion estimation
Similar to the RNA-seq data, the variability between biological replicates has also been observed in bisulfite sequencing data. This variability can be captured by the NB dispersion parameter under the generalized linear model (GLM) framework in EdgeR.
differentially methylated CpG loci Test is performed using likelihood ratio tests (LRT) in EdgeR.
> topTags(lrt)
Coefficient: 1*P6 -0.5*P7 -0.5*P8
Chr Locus EntrezID Symbol Strand Distance Width logFC
chr16-76326604 chr16 76326604 268903 Nrip1 - -46445 85647 -8.87
chr13-45709467 chr13 45709467 66355 Gmpr + -202023 38943 7.60
chr10-40387375 chr10 40387375 78334 Cdk19 + -38067 134511 -8.13
chr11-100144651 chr11 100144651 16669 Krt19 - 952 2889 -8.19
chr17-46572098 chr17 46572098 20807 Srf - 15936 9324 9.10
chr13-45709489 chr13 45709489 66355 Gmpr + -202045 38943 7.47
chr3-54724012 chr3 54724012 69639 Exosc8 - -11352 6686 -8.40
chr13-45709480 chr13 45709480 66355 Gmpr + -202036 38943 7.70
chr8-120068504 chr8 120068504 102193 Zdhhc7 - -32968 20378 7.42
chr2-69631013 chr2 69631013 72569 Bbs5 + 16242 20315 -7.30
logCPM LR PValue FDR
chr16-76326604 1.52 321 8.35e-72 4.27e-66
chr13-45709467 1.98 319 2.62e-71 6.70e-66
chr10-40387375 1.77 312 6.46e-70 1.10e-64
chr11-100144651 1.58 306 1.40e-68 1.79e-63
chr17-46572098 1.61 302 1.44e-67 1.48e-62
chr13-45709489 1.97 294 5.46e-66 4.66e-61
chr3-54724012 1.31 292 1.56e-65 1.14e-60
chr13-45709480 1.97 292 2.01e-65 1.28e-60
chr8-120068504 1.73 285 7.12e-64 4.05e-59
chr2-69631013 2.03 277 3.66e-62 1.88e-57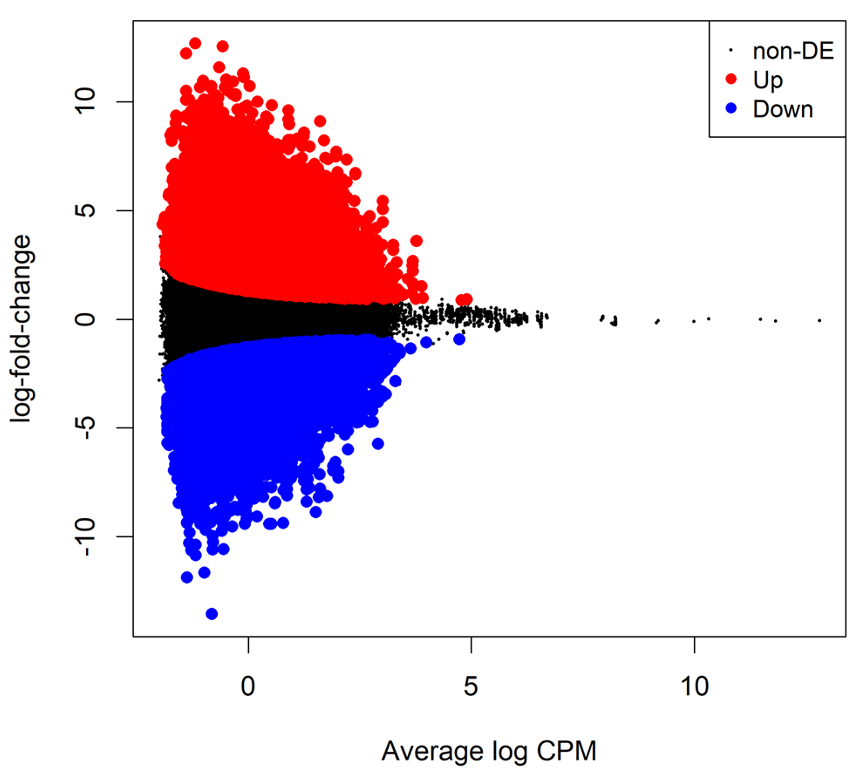
DAG of a complete DNA methylation data analysis pipeline
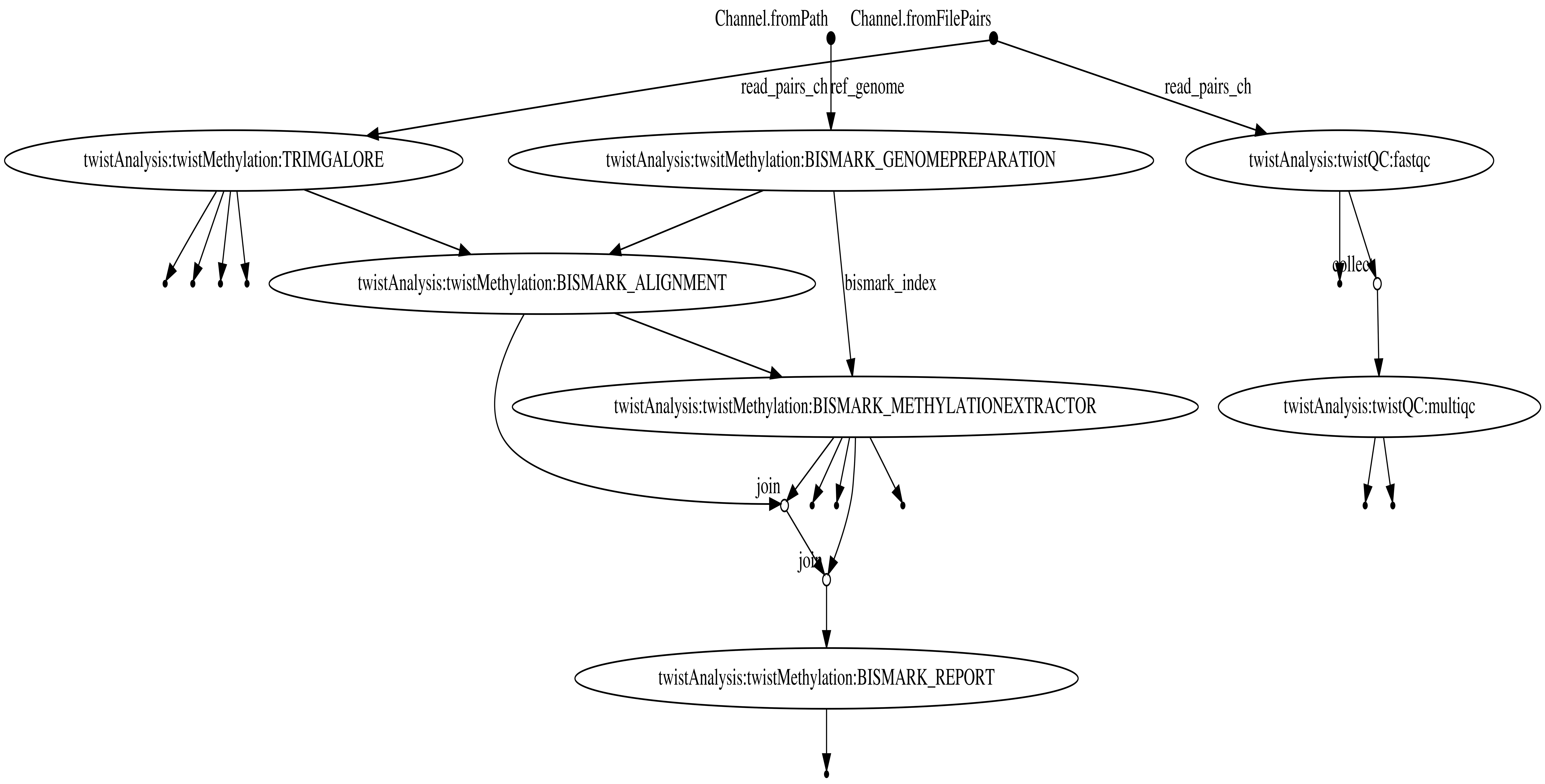

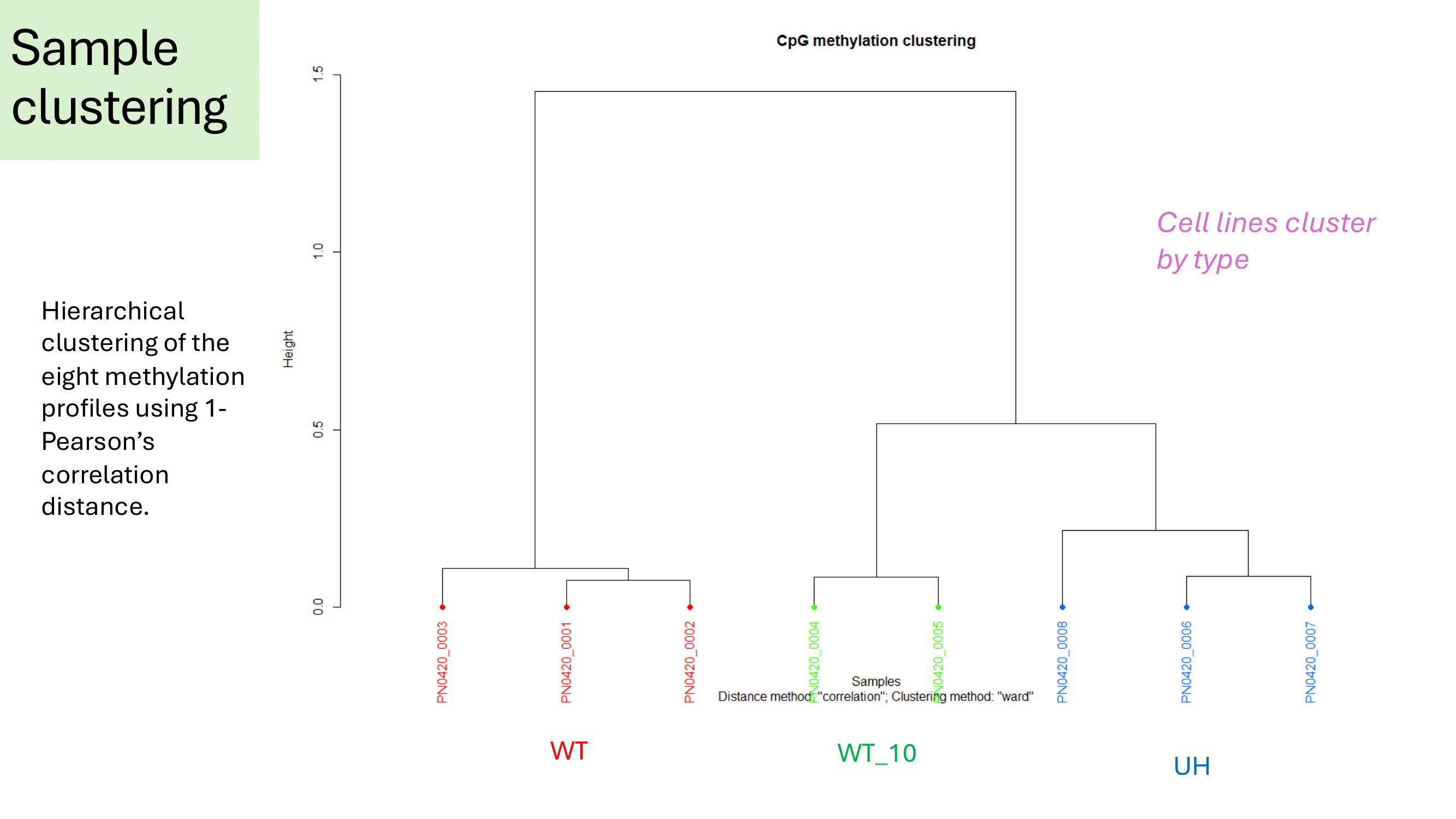
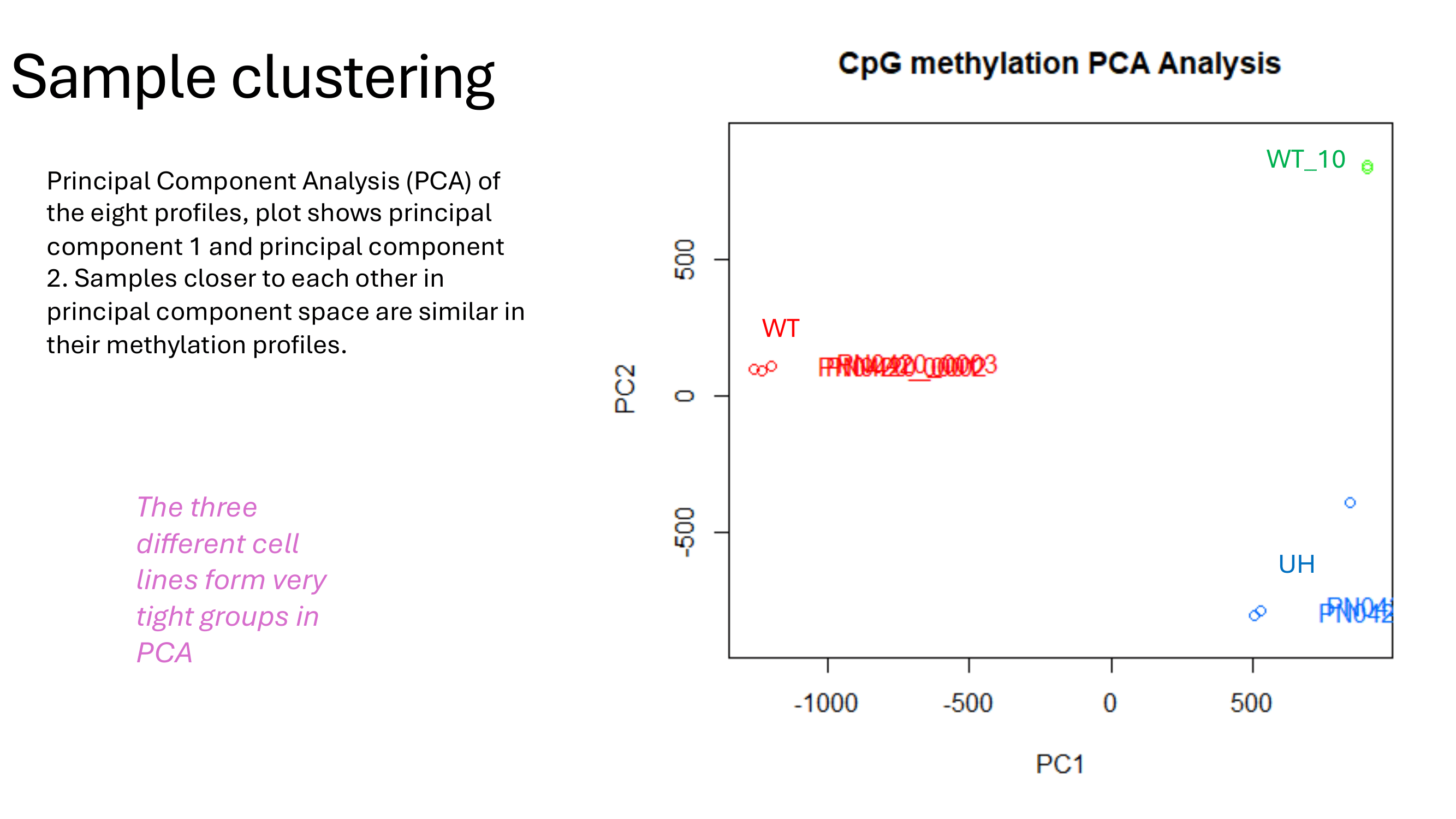
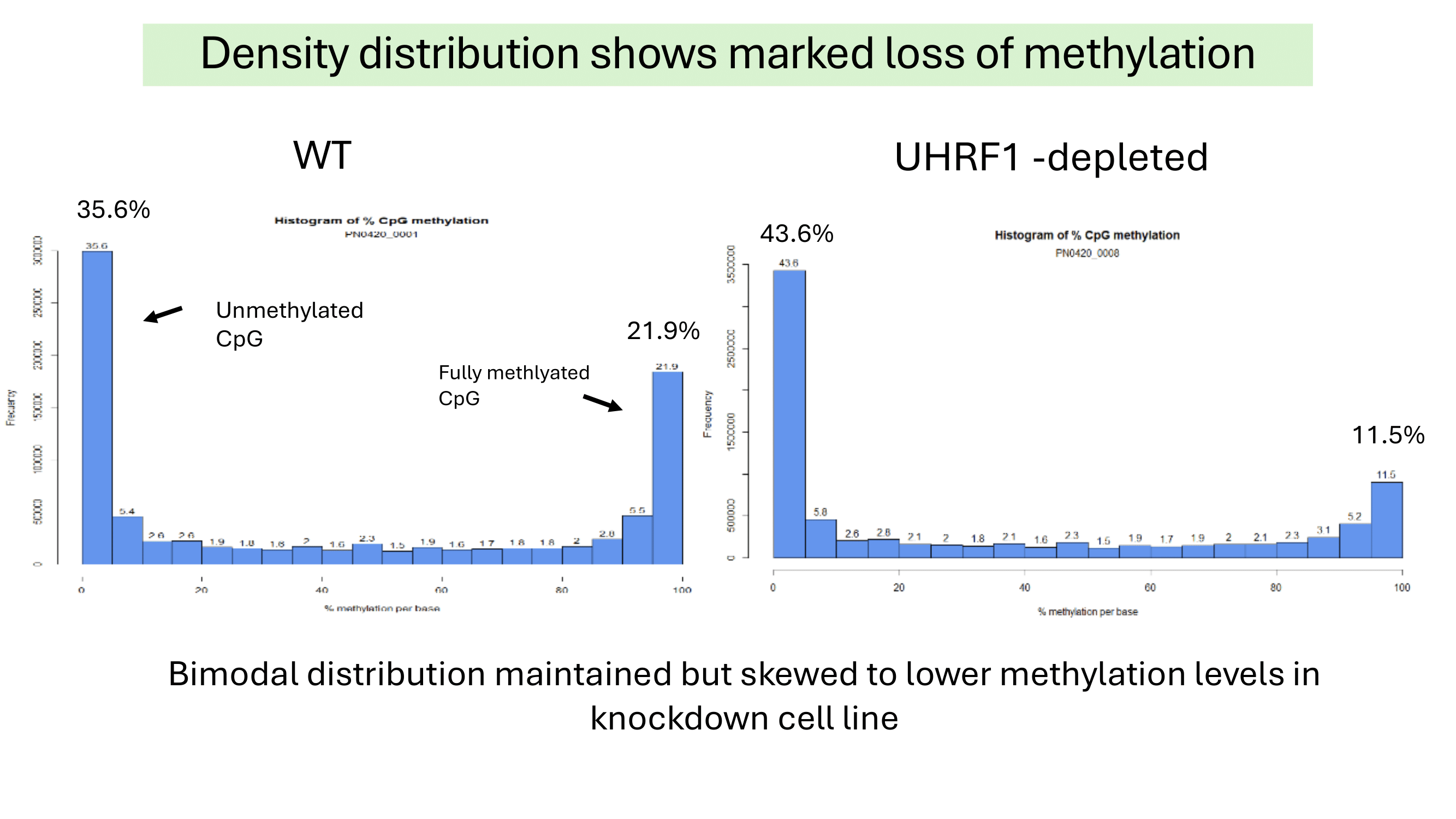

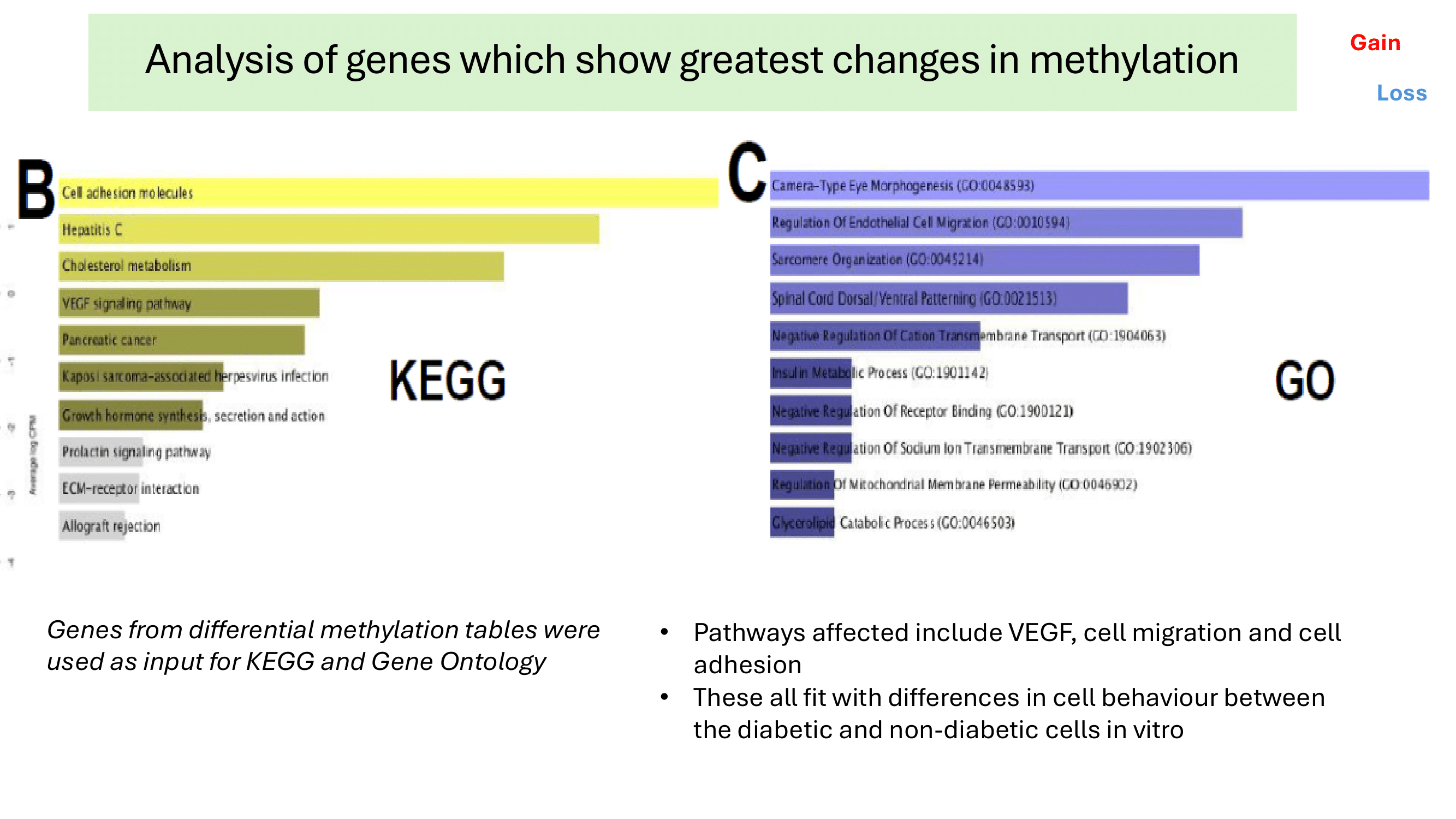
Summary
The pipeline is under-development. For updates, please visit https://github.com/JD2112/TwistNF/
The pipeline uses publicly available packages and tools for data analysis
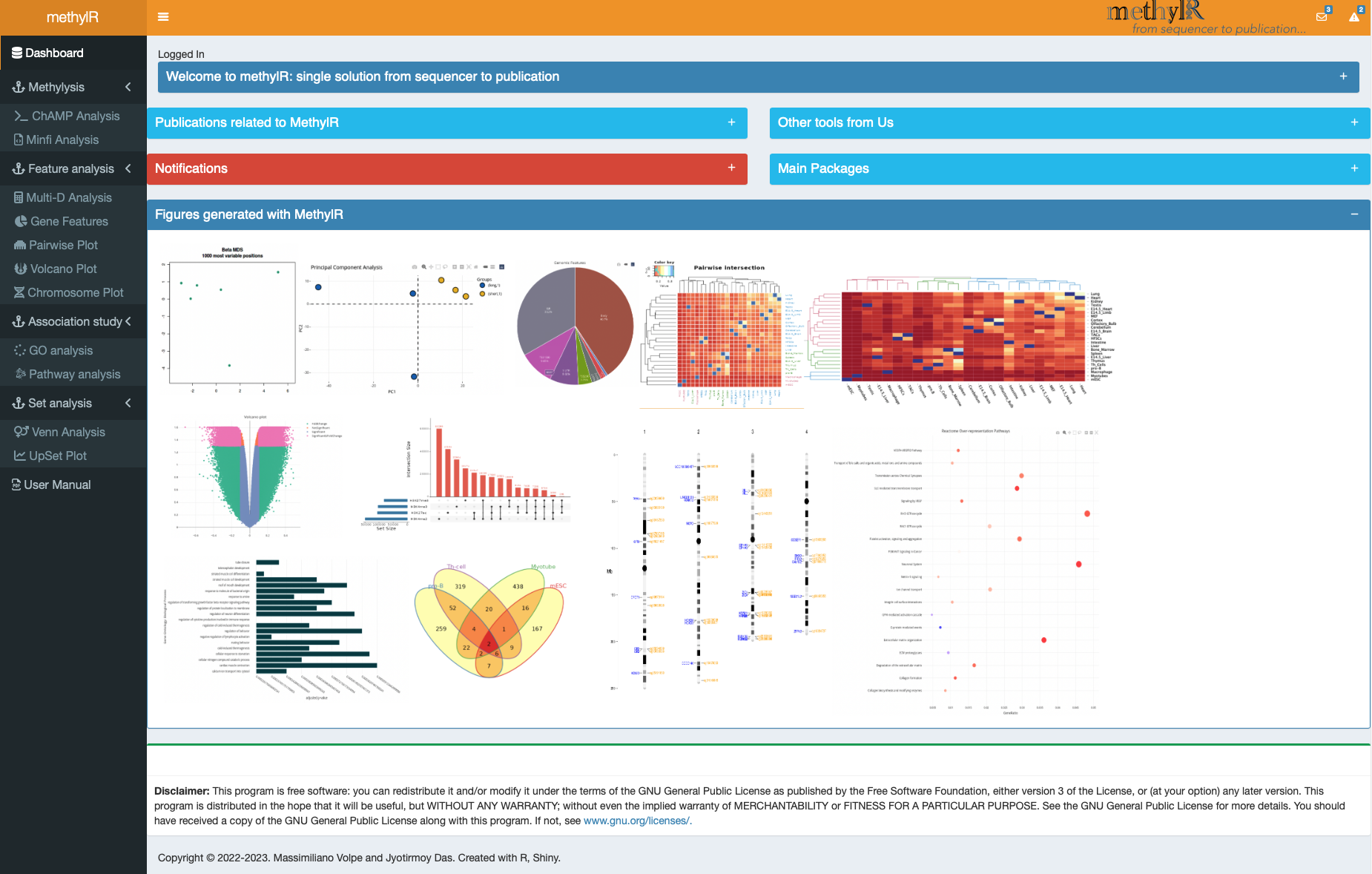
Further downstream analysis can be done on methylr
Special thanks to -
Colum Walsh
Malgorzata Lysiak
Esther González-Padilla
Vesa Loitto
Special thanks to -
Colum Walsh
Malgorzata Lysiak
Esther González-Padilla
Vesa Loitto


{kind=link}